Research
Advancing Non-adiabatic Quantum Dynamics
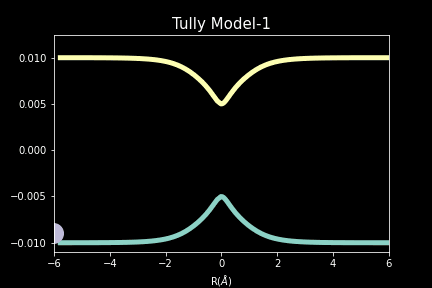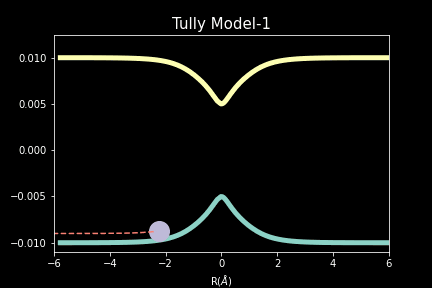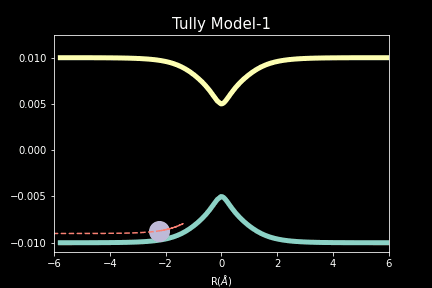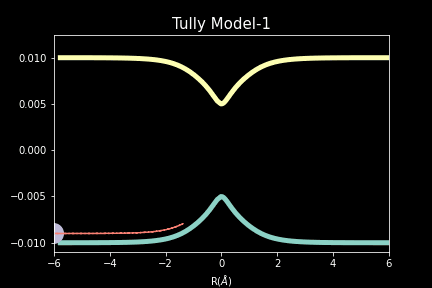
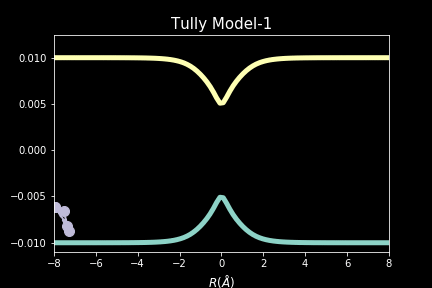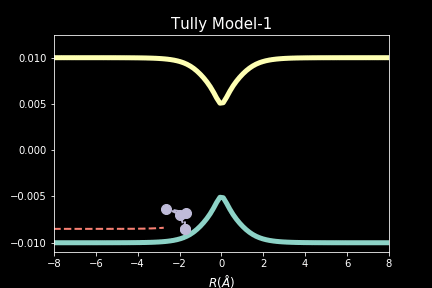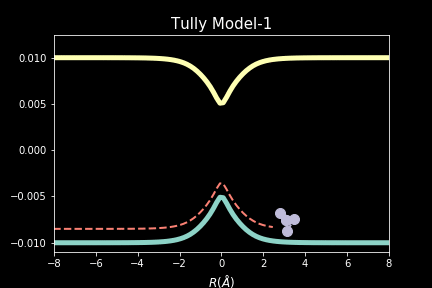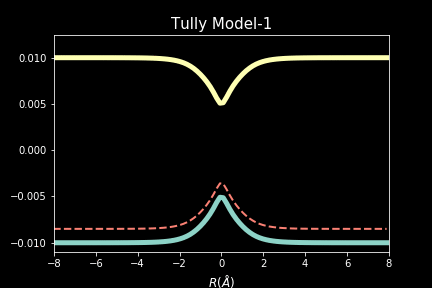
The study explores quantum-classical dynamics methodologies, focusing on the fewest-switches surface hopping (FSSH) as a revolutionary approach for efficiently studying charge transfer (CT) reactions. Despite the limitations of common trajectory-based methods like FSSH in incorporating nuclear quantum effects (NQEs), the project introduces the SHARP Pack (short for Surface Hopping And Ring Polymer package) software package incorporating ring polymer surface hopping (RPSH) method, combining FSSH and ring polymer molecular dynamics (RPMD) strengths. The developed modular software for RPSH aims to establish it as a mainstream methodology for nonadiabatic molecular dynamics simulations with a focus on NQEs. A comprehensive analysis evaluates RPSH's accuracy and limitations across various model systems a nd reaction regimes, highlighting its significant advancement in incorporating NQEs in nonadiabatic molecular dynamics. The exploration of velocity rescaling schemes and remedies for frustrated hops enhances RPSH's applicability and robustness in nonadiabatic simulations.
- Limbu, Shakib, J. Phys. Chem. Lett., 14, 8658-8666 (2023)
- Limbu, Shakib, Software Impacts, 19, 100604 (2024)
Disorder by Design: Application to Amorphous Semiconductors
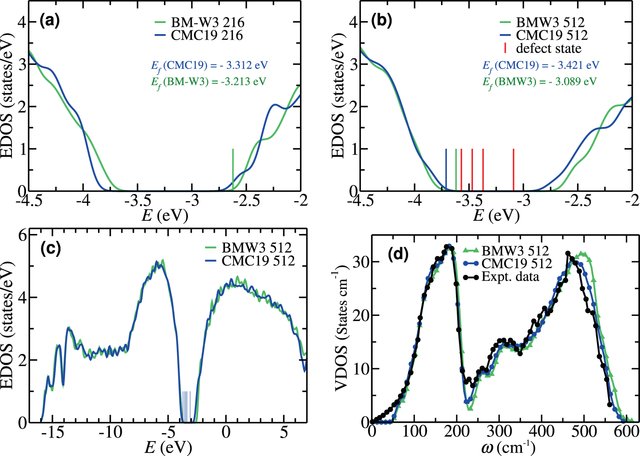
While conventional approaches to materials modeling made significant contributions and advanced our understanding of materials properties in the past decades, these approaches often cannot be applied to disordered materials for which accurate total-energy functionals or forces are either not available or it is simply infeasible to employ due to computational complexities associated with modeling disordered solids in the absence of translational symmetry. Here, we adopted an information-driven probabilistic viewpoint and addressed materials design as an optimization program, jointly supported by experimental data and information, by solving a long-standing difficult problem involving a structural determination of amorphous silicon via inversion of diffraction data. Even this approach can produce microstructural properties of realistic samples of a-Si from experiments, such as voids and vacancy-type defects, and demonstrates that data-driven approaches can enhance existing methodologies for modeling disordered materials based on an information paradigm. This brings a directional step-change in materials modeling computation.
- Limbu, Elliott, Atta-Fynn, Biswas*, Scientific Reports, 10, 7742 (2020)
Information-Driven Inverse Approach for Amorphous Silicon
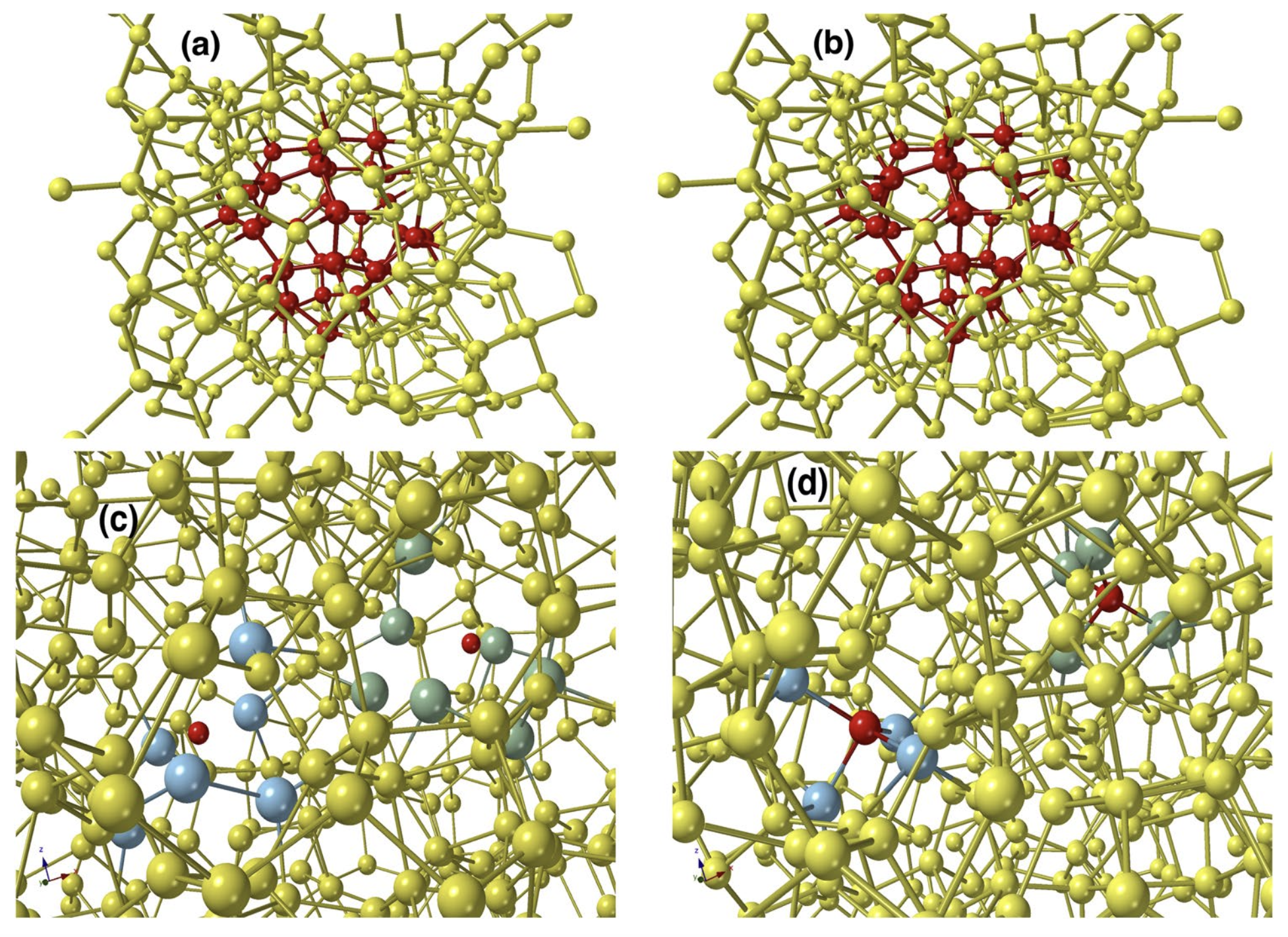
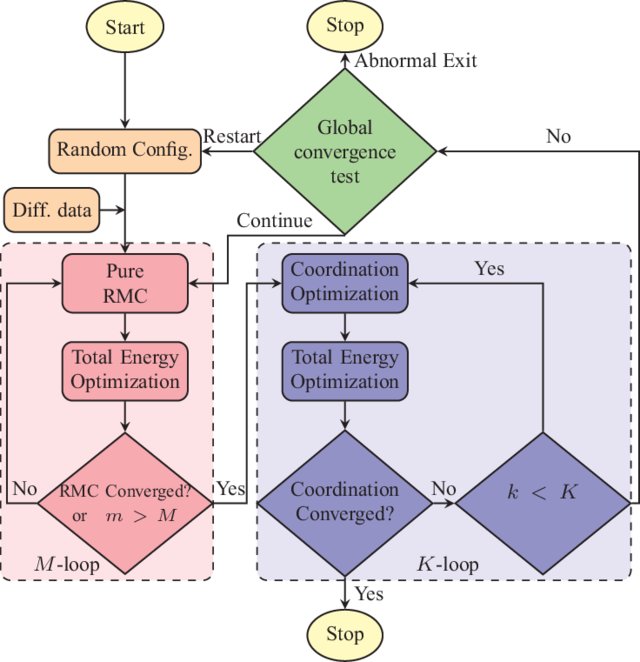
A very different approach to the materials-structure determination problem for amorphous solids is to address the problem from a hybrid point of view. Based on a hybrid scheme and incorporating experimental diffraction data, a few structural constraints, and a total-energy functional, we presented an accurate structural solution of the inverse problem by developing a new information-driven inverse approach. By introducing a subspace optimization technique, the difficulty associated with the optimization of the augmented objective function can be reduced considerably to determine optimal structural solutions, satisfying experiments, and a total-energy functional simultaneously. This method can produce nearly defect-free models of amorphous silicon which have structural, electronic, and vibrational properties fully consistent with experimental data. The realistic atomistic models of a-Si exhibit a clean gap around the Fermi level in the electronic spectrum.
- Limbu, Atta-Fynn, Drabold, Elliott, Biswas*, Phys. Rev. Materials, 2, 115602 (2018)
- Limbu*, Atta-Fynn, Biswas, MRS Adv., 4, 87-93 (2019)
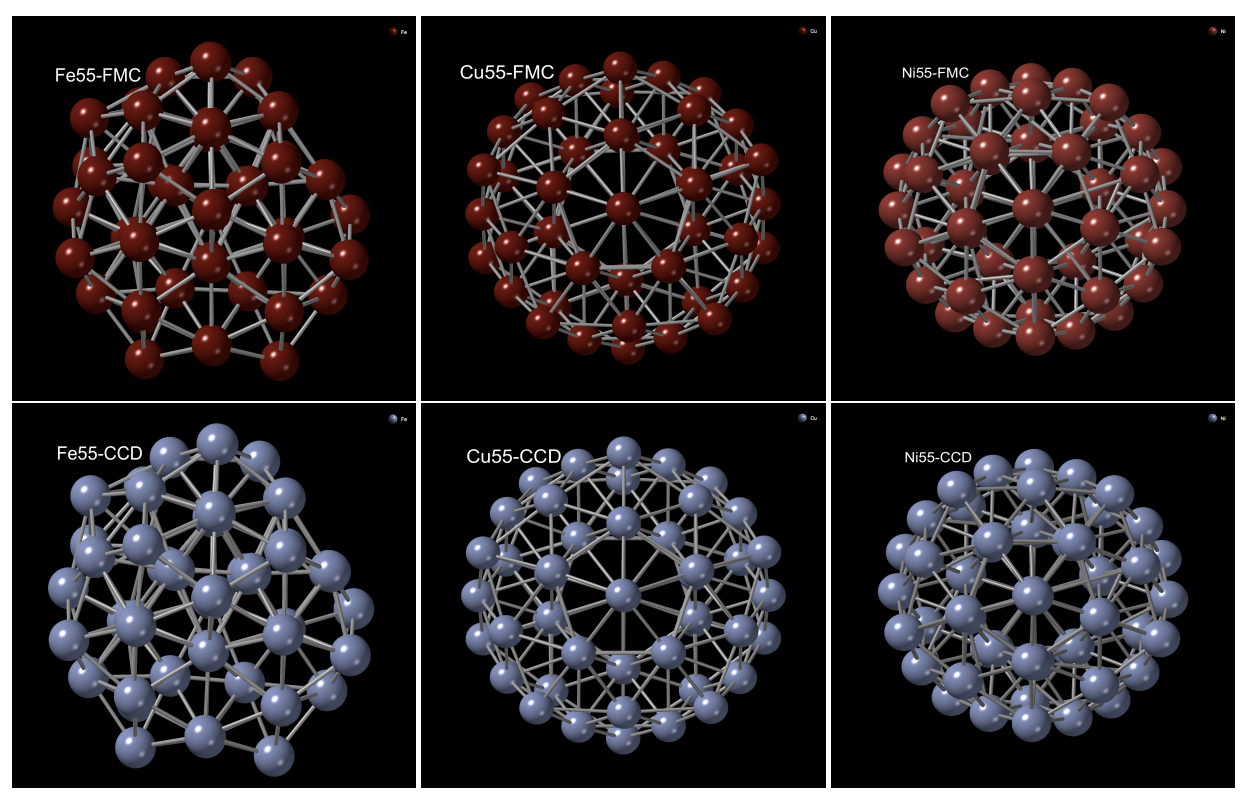
Force-biased Monte Carlo (FMC) and ab initio modeling of transition metal clusters
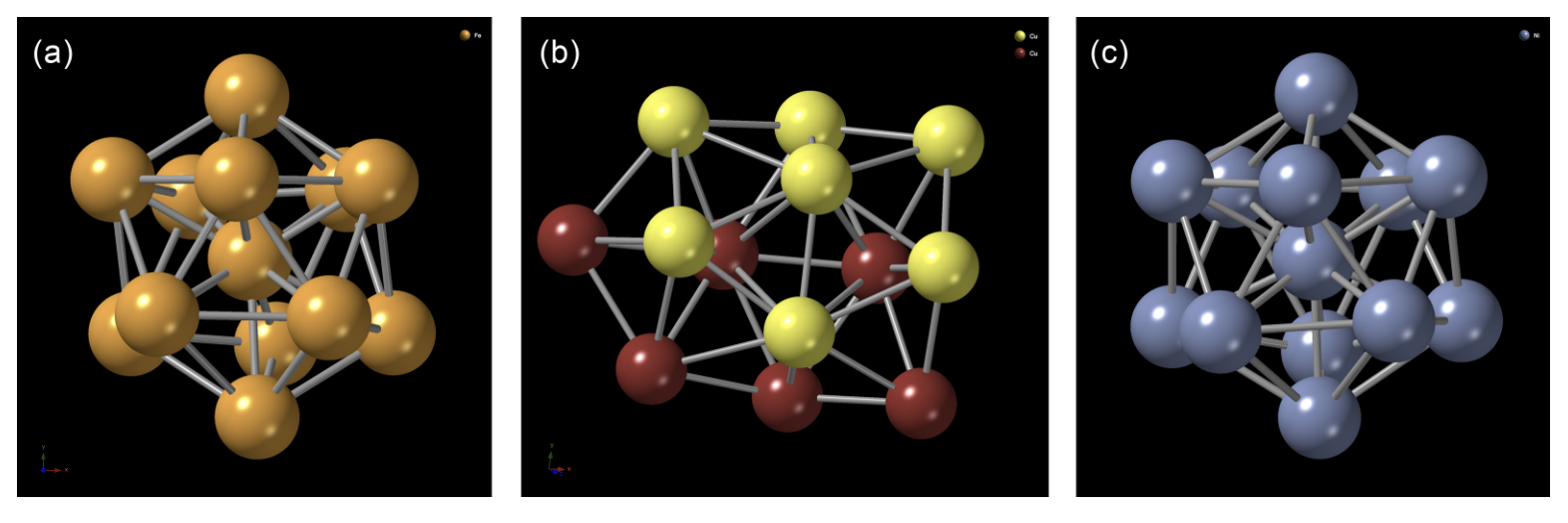
A force-biased Monte Carlo (FMC) method, based on the Finnis-Sinclair and the Sutton-Chen potentials, was used to model the putative ground-state structure of the transition-metal clusters (TMC) of Fe, Ni, and Cu with sizes of 1-60 atoms. Specifically, the total energy of the clusters was minimized using the local gradient of the potentials in Monte Carlo simulations. The atomic structure of the FMC clusters was analyzed and compared with their counterparts from the Cambridge Cluster Database (CCD) upon relaxation of the clusters with the planewave module in density-functional-theory code NWCHEM. An atom-by-atom comparison of the FMC and CCD clusters was conducted by superposing one set of clusters onto another, and the electronic properties of the clusters were addressed by computing the density of electronic states by constructing DFT Hamiltonians.
- Limbu, Biswas*, J. Phys.: Conf. Series, 921, 012010 (2017)
- Limbu, Atta-Fynn, Drabold, Elliot, Biswas*, Phys. Rev. B, 96, 174208 (2017)
- Limbu, Madueke, Atta-Fynn, Drabold, Biswas*, J. Phys.: Conf. Series, 1252, 012009 (2019)